


Drugs are introduced into the body by several routes. They may be
Taken by mouth (orally)
Given by injection into a vein (intravenously, IV), into a muscle (intramuscularly, IM), into the space around the spinal cord (intrathecally), or beneath the skin (subcutaneously, sc)
Placed under the tongue (sublingually) or between the gums and cheek (buccally)
Inserted in the rectum (rectally) or vagina (vaginally)
Placed in the eye (by the ocular route) or the ear (by the otic route)
Sprayed into the nose and absorbed through the nasal membranes (nasally)
Breathed into the lungs, usually through the mouth (by inhalation) or mouth and nose (by nebulization)
Applied to the skin (cutaneously) for a local (topical) or bodywide (systemic) effect
Delivered through the skin by a patch (transdermally) for a systemic effect
Each route has specific purposes, advantages, and disadvantages.
(See also Introduction to Administration and Kinetics of Drugs.)
Oral route
Many drugs can be administered orally as liquids, capsules, tablets, or chewable tablets. Because the oral route is the most convenient and usually the safest and least expensive, it is the one most often used. However, it has limitations because of the way a drug typically moves through the digestive tract. For drugs administered orally, absorption may begin in the mouth and stomach. However, most drugs are usually absorbed from the small intestine. The drug passes through the intestinal wall and travels to the liver before being transported via the bloodstream to its target site. The intestinal wall and liver chemically alter (metabolize) many drugs, decreasing the amount of drug reaching the bloodstream. Consequently, these drugs are often given in smaller doses when injected intravenously to produce the same effect.
When a drug is taken orally, food and other drugs in the digestive tract may affect how much of and how fast the drug is absorbed. Thus, some drugs should be taken on an empty stomach, others should be taken with food, others should not be taken with certain other drugs, and still others cannot be taken orally at all.
nonsteroidal anti-inflammatory drugs (NSAIDs) can harm the lining of the stomach and small intestine to potentially cause or aggravate preexisting ulcers. Other drugs are absorbed poorly or erratically in the digestive tract or are destroyed by the acid and digestive enzymes in the stomach.
Other routes of administration are required when the oral route cannot be used, for example:
When a person cannot take anything by mouth
When a drug must be administered rapidly or in a precise or very high dose
When a drug is poorly or erratically absorbed from the digestive tract
Injection routes
Administration by injection (parenteral administration) includes the following routes:
Subcutaneous (under the skin)
Intramuscular (in a muscle)
Intravenous (in a vein)
Intrathecal (around the spinal cord)
A drug product can be prepared or manufactured in ways that prolong drug absorption from the injection site for hours, days, or longer. Such products do not need to be administered as often as drug products with more rapid absorption.
Through the Skin
Sometimes a drug is given through the skin—by needle (subcutaneous, intramuscular, or intravenous route), by patch (transdermal route), or by implantation. |

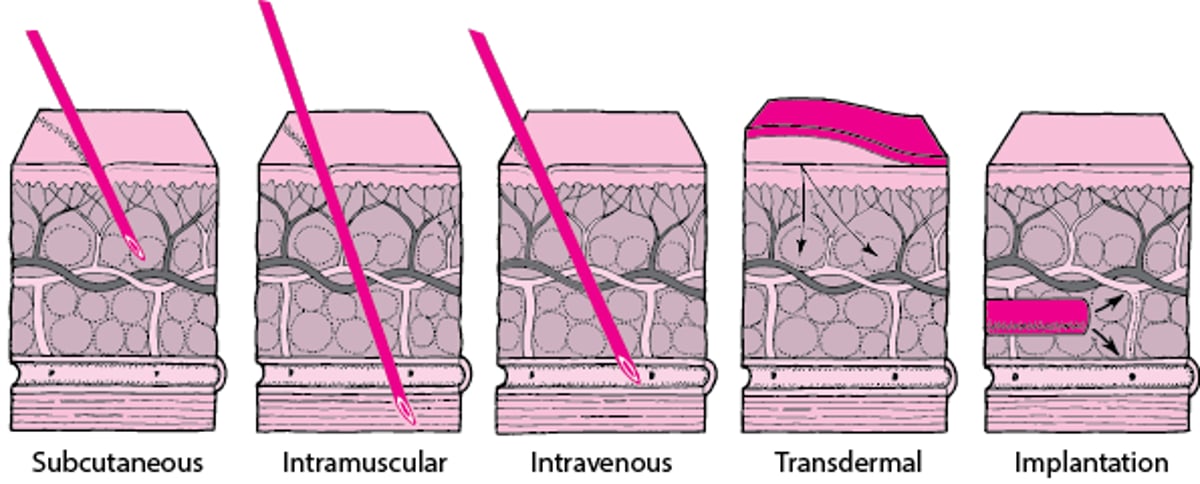
For the subcutaneous route, a needle is inserted into fatty tissue just beneath the skin. After a drug is injected, it then moves into small blood vessels (capillaries) and is carried away by the bloodstream. Alternatively, a drug reaches the bloodstream through the lymphatic vessels (see figure Lymphatic System: Helping Defend Against Infection
Certain drugs (such as progestins used for hormonal birth control
The intramuscular route is preferred to the subcutaneous route when larger volumes of a drug product are needed. Because the muscles lie below the skin and fatty tissues, a longer needle is used. Drugs are usually injected into the muscle of the upper arm, thigh, or buttock. How quickly the drug is absorbed into the bloodstream depends, in part, on the blood supply to the muscle: The sparser the blood supply, the longer it takes for the drug to be absorbed.
For the intravenous route, a needle is inserted directly into a vein. A solution containing the drug may be given in a single dose or by continuous infusion. For infusion, the solution is moved by gravity (from a collapsible plastic bag) or, more commonly, by an infusion pump through thin flexible tubing to a tube (catheter) inserted in a vein, usually in the forearm. Intravenous administration is the best way to deliver a precise dose quickly and in a well-controlled manner throughout the body. It is also used for irritating solutions, which would cause pain and damage tissues if given by subcutaneous or intramuscular injection. An intravenous injection can be more difficult to administer than a subcutaneous or intramuscular injection because inserting a needle or catheter into a vein may be difficult, especially if the person is obese.
When given intravenously, a drug is delivered immediately to the bloodstream and tends to take effect more quickly than when given by any other route. Consequently, health care practitioners closely monitor people who receive an intravenous injection for signs that the drug is working or is causing undesired side effects. Also, the effect of a drug given by this route tends to last for a shorter time. Therefore, some drugs must be given by continuous infusion to keep their effect constant.
For the intrathecal route,
Sublingual and buccal routes
Rectal route
Vaginal route
Some drugs may be administered vaginally to women as a solution, tablet, cream, gel, suppository, or ring. The drug is slowly absorbed through the vaginal wall. This route is often used to give estrogen to women during menopause to relieve vaginal symptoms such as dryness, soreness, and redness.
Ocular route
Drugs used to treat eye disorders (such as glaucoma, conjunctivitis, and injuries) can be mixed with inactive substances to make a liquid, gel, or ointment so that they can be applied to the eye. Liquid eye drops are relatively easy to use but may run off the eye too quickly to be absorbed well. Gel and ointment formulations keep the drug in contact with the eye surface longer, but they may blur vision. Solid inserts, which release the drug continuously and slowly, are also available, but they may be hard to put in and keep in place.
Ocular drugs are almost always used for their local effects. For example, artificial tears are used to relieve dry eyes. Other drugs (for example, those used to treat glaucoma [see table Drugs Used to Treat Glaucoma
Otic route
Nasal route
Inhalation route
Drugs administered by inhalation through the mouth must be atomized into smaller droplets than those administered by the nasal route, so that the drugs can pass through the windpipe (trachea) and into the lungs. How deeply into the lungs they go depends on the size of the droplets. Smaller droplets go deeper, which increases the amount of drug absorbed. Inside the lungs, they are absorbed into the bloodstream.
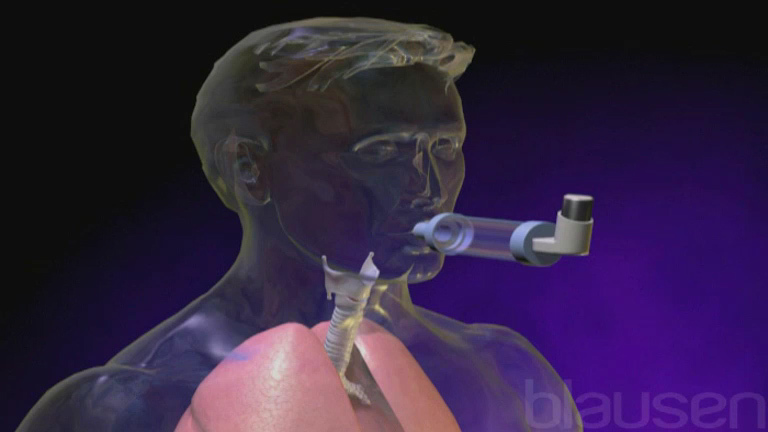

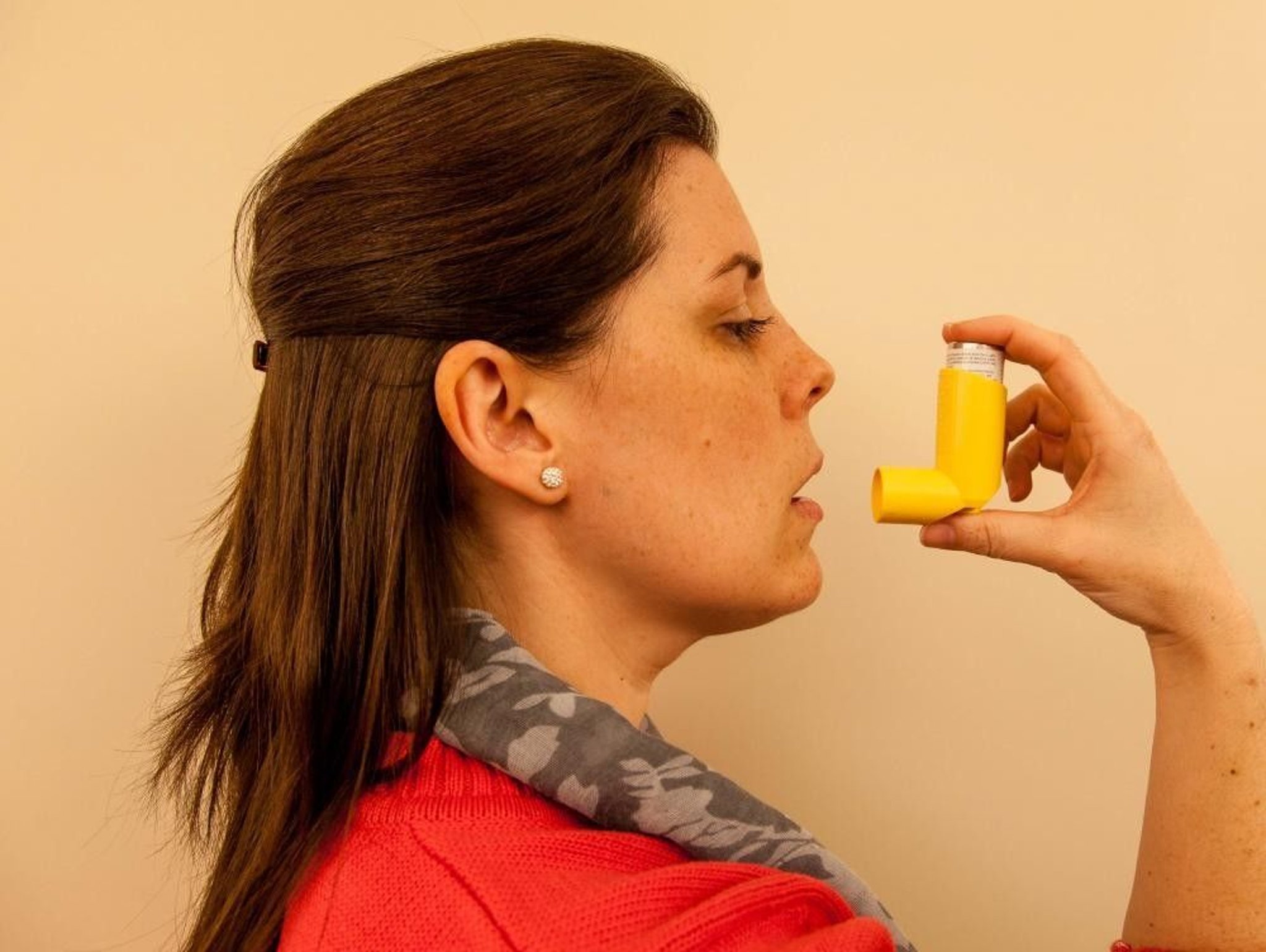
Photo courtesy of Justin Kaplan, MD.
Relatively few drugs are administered this way because inhalation must be carefully monitored to ensure that a person receives the right amount of drug within a specified time. In addition, specialized equipment may be needed to give the drug by this route. Usually, this method is used to administer drugs that act specifically on the lungs, such as aerosolized antiasthmatic drugs in metered-dose containers (called inhalers), and to administer gases used for general anesthesia.
Nebulization route
cystic fibrosispneumonia caused by Pneumocystis jiroveciiasthma attacks).
Side effects can include those that occur when the drug is deposited directly in the lungs (such as cough, wheezing, shortness of breath, and lung irritation), spread of the drug into the environment (possibly affecting people other than the one taking the drug), and contamination of the device used for nebulization (particularly when the device is reused and inadequately cleaned). Using the device properly helps prevent side effects.
Cutaneous route
Drugs applied to the skin are usually used for their local effects and thus are most commonly used to treat superficial skin disorders, such as psoriasis, eczema, skin infections (viral, bacterial, and fungal), itching, and dry skin. The drug is mixed with inactive substances. Depending on the consistency of the inactive substances, the formulation may be an ointment, cream, lotion, solution, powder, or gel (see Topical Preparations).






