


Twelve pairs of nerves—the cranial nerves—lead directly from the brain to various parts of the head, neck, and trunk. Some of the cranial nerves are involved in the special senses (such as seeing, hearing, and taste), and others control muscles in the face or regulate glands. The nerves are named and numbered (according to their location, from the front of the brain to the back).
Viewing the Cranial Nerves
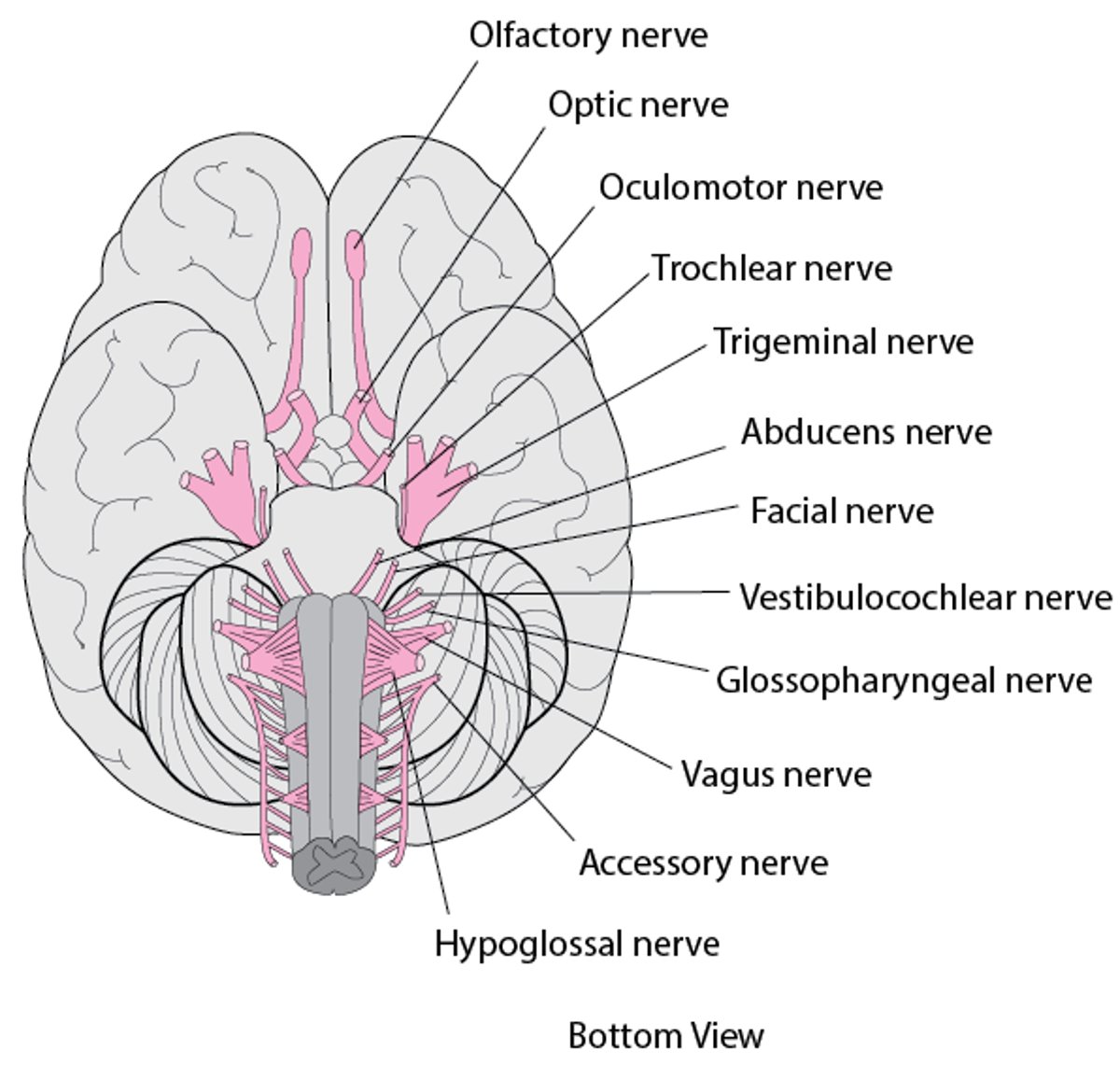
Twelve pairs of cranial nerves emerge from the underside of the brain, pass through openings in the skull, and lead to parts of the head, neck, and trunk. |

A cranial nerve disorder may result when the following are damaged or malfunction:
Areas of the brain that control cranial nerves (called centers, or nuclei), as may occur when a stroke damages the area that controls the facial nerve
The nerve fibers that connect cranial nerve centers within the brain, as occurs in internuclear ophthalmoplegia
Only one cranial nerve, as occurs in oculomotor palsy, trigeminal neuralgia, Bell palsy, and hemifacial spasm
Some cranial nerve disorders interfere with eye movement. Eye movement is controlled by 3 pairs of muscles. These muscles move the eye up and down, right and left, and diagonally. The muscles are controlled by the following cranial nerves:
If one of these nerves or the area in the brain that controls these muscles is damaged, the muscles may become paralyzed to varying degrees (called a palsy), and people may not be able to move their eyes normally. How eye movement is affected depends on which nerve is affected. People with one of these palsies may have double vision when they look in certain directions.
Did You Know...
|
Causes of Cranial Nerve Disorders
Cranial nerve disorders have many causes, including the following:
Head injuries
Tumors
Infections, such as COVID-19, Lyme disease, and shingles
An inadequate blood supply (as occurs in diabetes or stroke)
Pressure on a nerve due to abnormalities in blood vessel, such as a bulge (aneurysm) in an artery or an abnormal connection between an artery and a vein (arteriovenous malformation)
Disorders that cause nerve cells to degenerate, as occurs in amyotrophic lateral sclerosis (ALS) or multiple sclerosis
Disorders that cause inflammation of blood vessels (vasculitis), such as giant cell arteritis
Certain medications, particularly antibiotics such as aminoglycosides
Some toxins, such as mercury
Symptoms of Cranial Nerve Disorders
Symptoms of cranial nerve disorders depend on which nerves are damaged and how they were damaged. Cranial nerve disorders can affect smell, taste, vision, sensation in the face, facial expression, hearing, balance, speech, swallowing, and muscles of the neck.
For example, vision may be affected in various ways:
If one of the 2nd cranial nerves (optic nerve) is damaged, vision in the affected eye may be partially or completely lost.
If any of the three cranial nerves that control eye movement (3rd, 4th, or 6th cranial nerve) is damaged, people cannot move their eyes normally. Symptoms include double vision when looking in certain directions.
If the 3rd cranial nerve (oculomotor nerve) is paralyzed, people cannot raise their upper eyelid. It droops down over the eye and interferes with vision.
If the 8th cranial nerve (auditory or vestibulocochlear nerve) is damaged or malfunctions, people may have problems hearing and/or have vertigo—a feeling that they, their environment, or both are spinning.
Cranial nerve disorders can also cause various kinds of facial or head pain.
Diagnosis of Cranial Nerve Disorders
A doctor's evaluation
Tests of cranial nerve function
Usually magnetic resonance imaging
When doctors suspect a cranial nerve disorder, they ask the person detailed questions about the symptoms. They also test the function of the cranial nerves by asking the person to do simple tasks, such as to follow a moving target with the eyes.
Imaging of the brain with magnetic resonance imaging (MRI) is often needed.
Treatment of Cranial Nerve Disorders
Treatment of the cause
Treatment of specific cranial nerve disorders depends on the cause.






