


Cirrhosis is a late stage of hepatic fibrosis that has resulted in widespread distortion of normal hepatic architecture. Cirrhosis is characterized by regenerative nodules surrounded by dense fibrotic tissue. Symptoms may not develop for years and are often nonspecific (eg, anorexia, fatigue, weight loss). Late manifestations include portal hypertension, ascites, hepatic encephalopathy, and, when decompensation occurs, liver failure. Diagnosis is usually made using noninvasive imaging, although liver biopsy is required on rare occasions. Management involves supportive care and treatment of the causative liver disease.
Cirrhosis was the 16th leading cause of death worldwide in 2019 (1).
General reference
1. GBD 2019 Diseases and Injuries Collaborators: Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: A systematic analysis for the global Burden of Disease Study 2019. Lancet 396(10258):1204-1222. https://doi.org/10.1016/S0140-6736(20)30925-9
Etiology of Cirrhosis
The causes of cirrhosis are the same as those of fibrosis (see table Disorders and Medications/Substances That Can Cause Hepatic Fibrosis). In high-resource countries, most cases result from chronic alcohol use, chronic viral hepatitis (hepatitis B and C), or metabolic dysfunction–associated steatohepatitis (MASH, formerly known as nonalcoholic steatohepatitis/NASH). In parts of Asia and Africa, cirrhosis often results from endemic chronic hepatitis B. (See table Characteristics of Hepatitis Viruses for additional information on hepatitis B and C.) Cirrhosis of unknown etiology (cryptogenic cirrhosis) is becoming less common as many specific causes (eg, chronic hepatitis C, MASH) are identified. Injury to the bile ducts also can result in cirrhosis, as occurs in mechanical bile duct obstruction, primary biliary cholangitis, and primary sclerosing cholangitis.
Pathophysiology of Cirrhosis
There are 2 primary ingredients:
Hepatic fibrosis
Regenerating liver cells
In response to injury and loss, growth regulators induce hepatocellular hyperplasia (producing regenerating nodules) and arterial growth (angiogenesis). Among the growth regulators are cytokines and hepatic growth factors (eg, epithelial growth factor, hepatocyte growth factor, transforming growth factor-alpha, tumor necrosis factor). Insulin
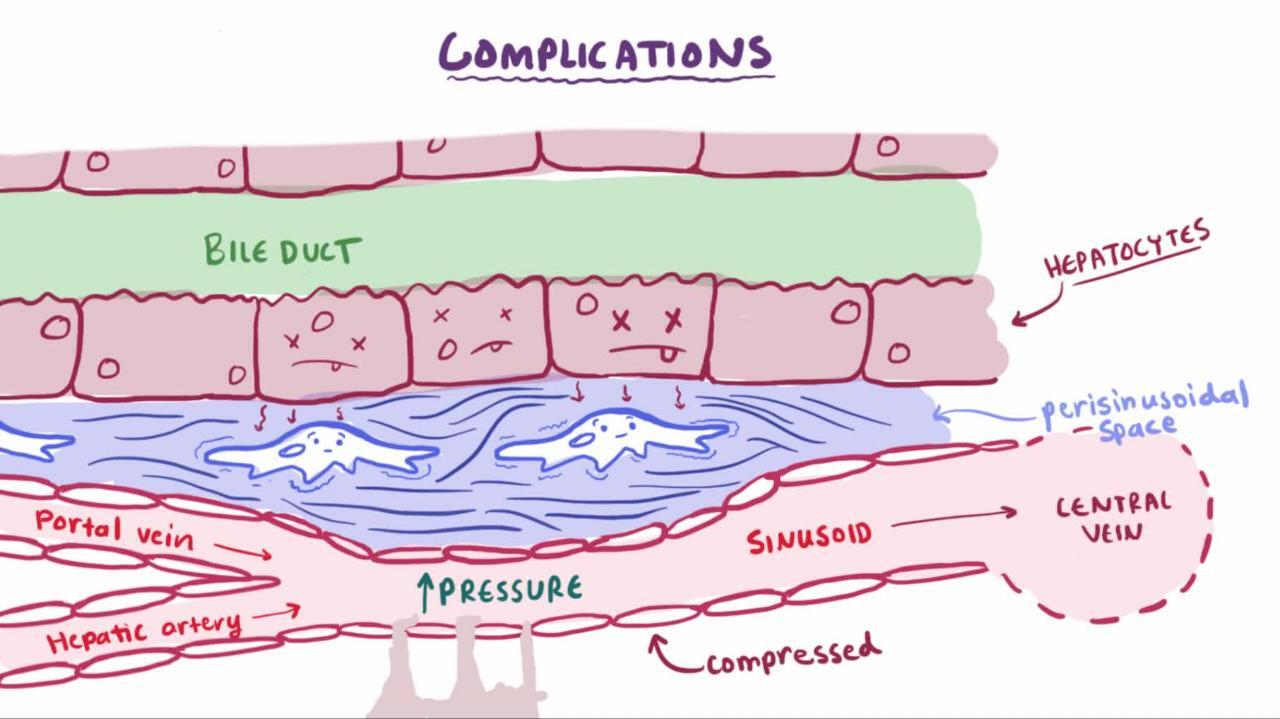
Angiogenesis produces new vessels within the fibrous sheath that surrounds nodules. These vessels connect the hepatic artery and portal vein to hepatic venules, restoring the intrahepatic circulatory pathways. Such interconnecting vessels provide relatively low-volume, high-pressure venous drainage that cannot accommodate as much blood volume as normal. As a result, portal vein pressure increases. Such distortions in blood flow contribute to portal hypertension, which increases because the regenerating nodules compress hepatic venules.
The progression rate from fibrosis to cirrhosis and the morphology of cirrhosis vary from person to person. Presumably, the reason for such variation is the extent of exposure to the injurious stimulus and the individual’s response.
Complications
Portal hypertension is the most common serious complication of cirrhosis, and it, in turn, causes complications, including
Gastrointestinal (GI) bleeding from esophageal, gastric, or rectal varices and portal hypertensive gastropathy
Acute kidney injury (hepatorenal syndrome)
Pulmonary hypertension (portopulmonary hypertension)
Hepatopulmonary syndrome (intrapulmonary shunt)
Ascites fluid can become infected (spontaneous bacterial peritonitis). Portopulmonary hypertension can manifest with symptoms of heart failure. Complications of portal hypertension tend to cause significant morbidity and mortality.
Cirrhosis can cause other cardiovascular complications. Vasodilation, intrapulmonary right-to-left shunting, and ventilation/perfusion mismatch can result in hypoxia (hepatopulmonary syndrome).
Progressive loss of hepatic architecture impairs function, leading to hepatic insufficiency; it manifests as coagulopathy, acute kidney injury (hepatorenal syndrome), and hepatic encephalopathy. Hepatic encephalopathy is characterized by asterixis, confusion, or hepatic coma and is the result of the liver's inability to metabolize the toxins from the gastrointestinal (GI) tract. Elevated serum ammonia level may help the diagnosis of hepatic encephalopathy, but the level does not correlate well with the severity of hepatic encephalopathy.
Hepatocytes secrete less bile, contributing to cholestasis and jaundicealcohol-related liver disease, from malabsorption due to pancreatic insufficiency.
Blood disorders are common. Anemia usually results from hypersplenism, chronic GI bleeding, folate deficiency (particularly in patients with alcohol use disorder), and hemolysis.
Cirrhosis results in decreased production of prothrombotic and antithrombotic factors. Hypersplenism and altered expression of thrombopoietin contribute to thrombocytopenia. Thrombocytopenia and decreased production of clotting factors can make clotting unpredictable, increasing risk of both bleeding and thromboembolic disease (even though international normalized ratio [INR] is usually increased). Leukopenia is also common; it is mediated by hypersplenism and altered expression of erythropoietin and granulocyte-stimulating factors.
Pearls & Pitfalls
|
Hepatocellular carcinoma frequently complicates cirrhosis from any cause (justifying clinical surveillance). The incidences of hepatocellular carcinoma in cirrhosis from specific etiologies are listed below (1):
Chronic hepatitis B: 3 to 8 % per year
Chronic hepatitis C: 3 to 5 % per year
Primary biliary cholangitis: 3 to 5 % per year
Metabolic dysfunction–associated steatohepatitis: 0.3 to 2.6% per year
Cirrhosis from other etiologies, eg, hemochromatosis, alcohol-related liver disease, alpha-1 antitrypsin deficiency, Wilson disease: Probably more than 1.5% per year
Histopathology
Cirrhosis is characterized by regenerating nodules and bridging fibrosis. Incompletely formed liver nodules, nodules without fibrosis (nodular regenerative hyperplasia), and congenital hepatic fibrosis (ie, widespread fibrosis without regenerating nodules) are not true cirrhosis.
Cirrhosis can be micronodular or macronodular. Micronodular cirrhosis is characterized by uniformly small nodules (< 3 mm in diameter) and thick regular bands of connective tissue. Typically, nodules lack lobular organization; terminal (central) hepatic venules and portal triads are distorted. With time, macronodular cirrhosis often develops. The nodules vary in size (3 mm to 5 cm in diameter) and have some relatively normal lobular organization of portal triads and terminal hepatic venules. Broad fibrous bands of varying thickness surround the large nodules. Collapse of the normal hepatic architecture is suggested by the concentration of portal triads within the fibrous scars. Mixed cirrhosis (incomplete septal cirrhosis) combines elements of micronodular and macronodular cirrhosis. Differentiation between these morphologic types of cirrhosis has limited clinical value.
Pathophysiology reference
1. Huang DQ, El-Serag HB, Loomba R: Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 8(4):223-238, 2021. doi: 10.1038/s41575-020-00381-6
Symptoms and Signs of Cirrhosis
Cirrhosis may be asymptomatic for years as long as it is compensated. Often, the first symptoms are nonspecific; they include generalized fatigue (due to cytokine release), anorexia, malaise, and weight loss (see table Common Symptoms and Signs Due to Complications of Cirrhosis). The liver is typically palpable and firm, with a blunt edge, but is sometimes small and difficult to palpate. Nodules usually are not palpable.
Clinical signs that suggest a chronic liver disorder or chronic alcohol use but are not specific for cirrhosis include muscle wasting, palmar erythema, parotid gland enlargement, white nails, clubbing, Dupuytren contracture, spider angiomas, gynecomastia, axillary hair loss, testicular atrophy, and peripheral neuropathy.
Once any complication of cirrhosis develops, additional decompensation (characterized by gastrointestinal bleeding, ascites, or hepatic encephalopathy) is much more likely.
Diagnosis of Cirrhosis
Liver blood tests, coagulation tests, complete blood count (CBC), and serologic tests for viral causes
Conventional liver imaging tests: Ultrasonography, CT, MRI
Noninvasive imaging assessment of fibrosis: Transient elastography, acoustic radiation force impulse imaging, 2-dimensional shear wave elastography, magnetic resonance elastography +/- proton density fat fraction
Identification of cause based on clinical evaluation, routine testing for common causes, and selective testing for less common causes
Sometimes liver biopsy (eg, when clinical and noninvasive tests are inconclusive, or when biopsy results may change management)
General approach
Cirrhosis is suspected in patients with manifestations of any of its complications (see table Common Symptoms and Signs Due to Complications of Cirrhosis), particularly portal hypertension or ascites. Early cirrhosis should be considered in patients with nonspecific symptoms or characteristic laboratory abnormalities detected incidentally during laboratory testing, particularly in patients who have a disorder or take a medication that might cause fibrosis.
Testing seeks to detect cirrhosis and any complications and to determine its cause.
Laboratory tests
Diagnostic testing begins with liver blood tests, coagulation tests, CBC, and serologic tests for chronic viral hepatitis (see tables Hepatitis B Serology and Hepatitis C Serology). Laboratory tests alone may increase suspicion for cirrhosis but cannot confirm or exclude it.
Test results may be normal or may indicate nonspecific abnormalities. Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels are often modestly elevated, but they can be normal. Alkaline phosphatase and gamma-glutamyl transpeptidase (GGT) are often normal; elevated levels indicate cholestasis or biliary obstruction. Bilirubin is usually normal but increases when cirrhosis progresses, particularly in primary biliary cholangitis. Decreased serum albumin and a prolonged prothrombin time (PT) directly reflect impaired hepatic synthesis—usually an end-stage event. Albumin can also be low when nutrition is poor.
Anemia is common and usually normocytic with a high red blood cell distribution width (RDW). Anemia is often multifactorial; contributing factors may include chronic gastrointestinal bleeding (usually causing microcytic anemia), folate deficiency (causing macrocytic anemia, especially in chronic excessive alcohol use), hemolysis, and hypersplenism. CBC may also detect leukopenia, thrombocytopenia, or pancytopenia.
Diagnostic imaging
Conventional imaging tests are not highly sensitive or specific for the diagnosis of cirrhosis by themselves, but they can often detect its complications. Noninvasive imaging studies (eg, transient elastography, acoustic radiation force impulse imaging, 2-dimensional shear wave elastography, and magnetic resonance elastography) are useful in detection of early cirrhosis when conventional imaging findings are equivocal and portal hypertension is not evident.
In advanced cirrhosis, ultrasonography shows a small, nodular liver. Ultrasonography also detects portal hypertension and ascites.
CT and MRI with and without contrast can detect a nodular texture, varices, portal/splenic vein thrombosis, and delineate a liver lesion suspect for hepatocellular carcinoma. Radionuclide liver scans using technetium-99m sulfur colloid may show irregular liver uptake and increased spleen and bone marrow uptake, but it has limited use in contemporary practice.
Identification of the cause
Determining the specific cause of cirrhosis requires key clinical information from the history and examination, as well as selective testing.
Alcohol is the likely cause in patients with a documented history of alcohol use disorder and laboratory findings of AST higher than ALT (especially AST/ALT ratio > 2), elevated gamma-glutamyl transpeptidase (GGT), and . Fever, tender hepatomegaly, and jaundice suggest the presence of acute alcoholic hepatitis.
Detecting serum antibody to hepatitis C (anti-HCV) and HCV-RNA indicates hepatitis C. Detecting hepatitis B surface antigen (HBsAg) and hepatitis B core antibody (anti-HBcAb) confirms chronic hepatitis B. Chronic hepatitis B with very low HBV viral load can occur in HBV/HDV co-infection. Most clinicians also routinely test for the following:
Autoimmune hepatitis: Suggested by a high antinuclear antibody titer (a low titer is nonspecific and does not always mandate further evaluation) and confirmed by hypergammaglobulinemia (IgG) and the presence of other autoantibodies (eg, anti–smooth muscle or anti-liver/kidney microsomal type 1 antibodies)
Hemochromatosis: Suggested by increased serum iron and transferrin saturation and confirmed by homeostatic iron regulator (HFE) genetic testing
Alpha-1 antitrypsin deficiency: Suggested by a low serum alpha-1 antitrypsin level and confirmed by genotyping/phenotyping
If these causes are not confirmed, other causes are sought:
Presence of antimitochondrial antibodies (in 95%) and elevated IgM suggest primary biliary cholangitis (PBC), which needs to be confirmed with biopsy.
Strictures and dilations of the intrahepatic and extrahepatic bile ducts, seen on magnetic resonance cholangiopancreatography (MRCP), suggest primary sclerosing cholangitis (PSC).
Decreased serum ceruloplasmin and characteristic copper test results suggest Wilson disease.
The presence of obesity and a history of diabetes suggest metabolic dysfunction–associated steatohepatitis (MASH), a diagnosis of exclusion unless confirmed with liver biopsy.
Liver biopsy
Liver biopsy is invasive and is subject to sampling error, but it remains the gold standard for the diagnosis of cirrhosis. Liver biopsy is required in the following situations:
If clinical criteria and noninvasive testing are inconclusive for diagnosis of cirrhosis or its etiology (for example, if well-compensated cirrhosis is suspected clinically and imaging findings are inconclusive)
To confirm certain causes of cirrhosis (eg, amyloidosis, PBC, or small duct PSC)
To assess the severity and/or activity of some causes of cirrhosis (eg, autoimmune hepatitis) in order to direct the intensity of treatment.
To confirm cirrhosis for certain disorders for which noninvasive imaging for fibrosis assessment has not been validated (eg, pregnancy, congestive hepatopathy, and rare liver disorders)
In obvious cases of cirrhosis with marked coagulopathy, portal hypertension, ascites, and liver failure, biopsy is not required unless results would change management. In patients with ascites, coagulopathy, and thrombocytopenia, the transjugular approach to biopsy is safest. When this approach is used, pressures can be measured and thus the trans-sinusoidal pressure gradient can be calculated.
Monitoring
All patients with cirrhosis, regardless of cause, should be screened regularly for hepatocellular carcinoma. Abdominal ultrasonography with or without serum alpha-fetoprotein (AFP) is recommended every 6 months. If abnormalities that are suspect for HCC are detected, contrast-enhanced MRI or triple-phase CT of the abdomen (pre-contrast, arterial phase, and venous phase) should be done. Certain features on contrast imaging (Liver Imaging Reporting and Data System 5 criteria, including early arterial enhancement, washout in portal phase, enhancing capsule) can confirm HCC, sparing the patient a biopsy. Contrast-enhanced ultrasonography appears promising as an alternative to CT or MRI but is still under study.
The presence of clinically significant portal hypertension (defined as hepatic venous pressure gradient ≥ 10 mmHg) should be assessed in patients with compensated cirrhosis, in a manner consistent with the Baveno VII consensus guidelines (1). Proposed diagnostic criteria using noninvasive methods to diagnose clinically significant portal hypertension include the following (2):
Liver stiffness (LS) on transient elastography (TE) ≥ 25 kPa
LS on TE 20 – 25 kPa and platelet count <150 k/mL
LS on TE 15 – 20 kPa and platelet count <110 k/mL
Treatment of Cirrhosis.) Upper endoscopy is not required to screen for gastroesophageal varices in patients on nonselective beta-blockers with compensated cirrhosis. However, if the patient is not a candidate for a nonselective beta-blocker, an upper endoscopy should be done every 2 to 3 years to monitor for gastroesophageal varices, and endoscopic banding should be performed as needed.
Diagnosis references
1. de Franchis R, Bosch J, Garcia-Tsao G, Baveno VII Faculty.: Baveno VII - Renewing consensus in portal hypertension. J Hepatol 76(4):959-974, 2022. doi: 10.1016/j.jhep.2021.12.022. Erratum in: J Hepatol. 2022 Apr 14
2. Podrug K, Trkulja V, Zelenika M, et al: Validation of the new diagnostic criteria for clinically significant portal hypertension by platelets and elastography. Dig Dis Sci 67(7):3327-3332, 2022. doi: 10.1007/s10620-021-07277-8
Treatment of Cirrhosis
Supportive care
In general, treatment is supportive and includes stopping injurious medications, providing nutrition (including supplemental vitamins), and treating the underlying disorders and complications. Doses of medications metabolized in the liver should be reduced. All alcohol and hepatotoxic substances must be avoided. Withdrawal symptoms during hospitalization should be anticipated in patients who have cirrhosis and have continued their chronic use of alcohol. Patients should be vaccinated against viral hepatitis A and B unless they are already immune.
Patients with varices need therapy to prevent bleeding (see Portal Hypertension: Treatment). Patients with compensated cirrhosis who have clinically significant portal hypertension (according to the Baveno VII criteria) should be started on a nonselective beta-blocker to prevent decompensation (1
Transjugular intrahepatic portosystemic shunting (TIPS) should be considered if patients have complications of portal hypertension that are refractory to standard treatments, including ascites and recurrent variceal bleeding. TIPS is relatively contraindicated in patients with heart failure, moderate or severe pulmonary hypertension, or hepatic encephalopathy. Patients with high MELD scores (> 18) have a higher risk of mortality after TIPS.
Liver transplantation is indicated for patients with end-stage liver disease or HCC. Risk of death without liver transplantation begins to exceed risks of transplantation (eg, perioperative complications, chronic immunosuppression) when the MELD score is more than about 15. Thus, if the score is ≥ 15, if the patient's HCC meets the criteria for MELD exception point, or if cirrhosis has decompensated clinically, patients should be referred to a transplantation center.
Treatment reference
1. de Franchis R, Bosch J, Garcia-Tsao G, Baveno VII Faculty.: Baveno VII - Renewing consensus in portal hypertension. J Hepatol 76(4):959-974, 2022. doi: 10.1016/j.jhep.2021.12.022. Epub 2021 Dec 30. Erratum in: J Hepatol. 2022 Apr 14.
Prognosis for Cirrhosis
Prognosis is often unpredictable. It depends on factors such as etiology, severity, presence of complications, comorbid conditions, host factors, and effectiveness of therapy. Cirrhosis was considered irreversible, but more recent evidence suggests it is reversible. Patients who continue to drink alcohol, even small amounts, have a very poor prognosis.
Child-Turcotte-Pugh classification for severity of liver disease
The Child-Turcotte-Pugh scoring system uses clinical and laboratory information to stratify disease severity, surgical risk, and overall prognosis (see tables Child-Turcotte-Pugh Scoring System and Interpretation of the Child-Turcotte-Pugh Scoring System). The Child-Turcotte-Pugh scoring system does, however, have limitations; for example, assessments of the severity of ascites and encephalopathy are subjective; inter-rater reliability of results is thus decreased.
Model for end-stage liver disease (MELD)
In contrast to the Child-Turcotte-Pugh classification, the model for end-stage liver disease (MELD) score estimates the severity of end-stage liver disease, regardless of cause, based solely on objective results of laboratory tests: serum creatinine, serum total bilirubin, and international normalized ratio (INR). The MELD score is used to determine allocation of available organs to liver transplant candidates because it can sort candidates by mortality risk (see table MELD Score and Mortality).
Variations of the MELD score are sometimes used for other purposes (eg, to estimate risk of 90-day mortality in patients with alcoholic hepatitis, to predict risk of postoperative mortality in patients with cirrhosis). A variation of the MELD score that incorporates serum sodium measurement (MELD-Na) more accurately predicts mortality in cirrhotic patients than the conventional MELD score, and was used by the United Network for Organ Sharing (UNOS)/Organ Procurement and Transplantation Network (OPTN) until July 2023 to prioritize patients on the liver transplant waiting list. Another variation of the MELD score (MELD 3.0) incorporated serum albumin/sex and updated the coefficients of existing variables, introduced interaction terms, and lowered the maximum creatinine to 3.0 mg/dL (1). In July 2023, UNOS/OPTN switched to using MELD 3.0 to prioritize liver transplant candidates age 12 years and older.
In 2019, the United Network for Organ Sharing (UNOS) implemented a major policy update on how the MELD exception (eg, HCC, hepatopulmonary syndrome) is handled. Under the new policy, patients are awarded a fixed MELD 3.0 score based on the Median MELD at Transplant (MMaT) in their region (which has a radius of 250 nautical miles), regardless of their waiting time.
Pediatric end-stage liver disease (PELD) score
For patients < 12 years, the corresponding pediatric end-stage liver disease (PELD) score is calculated. Higher PELD scores predict higher risk. In July 2023, UNOS/OPTN adopted a new variant of PELD score (PELD Cr), which incorporates creatinine, updated coefficients for existing variables, and the conversions of age and growth failure as continuous variables.
Treatment reference
1. Kim WR, Mannalithara A, Heimbach JK, et al: MELD 3.0: The model for end-stage liver disease updated for the modern era. Gastroenterology161(6):1887-1895.e4, 2021. doi: 10.1053/j.gastro.2021.08.050
Key Points
Morbidity and mortality in cirrhosis usually result from its complications (eg, complications of portal hypertension, liver failure, hematologic problems).
Do liver biopsy if a clear diagnosis would lead to better management and outcome.
Evaluate all patients with cirrhosis for autoimmune hepatitis, hereditary hemochromatosis, and alpha-1 antitrypsin deficiency, as well as for the more common causes, metabolic dysfunction-associated steatotic liver disease (MASLD, formerly known as nonalcoholic fatty liver disease/NAFLD), and alcoholic and viral hepatitis.
Monitor all patients periodically for clinically significant portal hypertension/gastroesophageal varices and hepatocellular carcinoma, doing testing as clinically indicated.
Predict prognosis using the Child-Turcotte-Pugh and model of end-stage liver disease (MELD) scoring systems, and refer patients with a MELD score ≥ 15 to be evaluated for a liver transplant.
Treat cirrhosis supportively, including using therapies to prevent bleeding.






