


Bronchiectasis is dilation and destruction of larger bronchi caused by chronic infection and inflammation. Common causes are cystic fibrosis, immune defects, and recurrent infections, though some cases seem to be idiopathic. Common symptoms are chronic cough and purulent sputum expectoration with or without dyspnea. Symptoms may worsen and may include fever during acute exacerbations. Diagnosis is based on history and imaging, usually involving high-resolution computed tomography, though standard chest x-rays may be diagnostic. Treatment and prevention of acute exacerbations are with bronchodilators, clearance of secretions, antibiotics, and management of complications, such as hemoptysis and further lung damage due to resistant or opportunistic infections. Treatment of underlying disorders is important whenever possible.
Etiology of Bronchiectasis
Bronchiectasis is best considered the common end-point of various disorders that cause chronic airway inflammation. Bronchiectasis may be
Diffuse: Affecting many areas of the lungs
Focal: Appearing in only 1 or 2 lung areas
Diffuse bronchiectasis
Diffuse bronchiectasis develops most often in patients with genetic, immunologic, or anatomic defects that affect the airways. The cause of many cases appears initially to be idiopathic, probably partly because onset is so slow that the triggering problem is not readily evident at the time bronchiectasis is recognized. However, after through investigation using newer genetic and immunologic testing,, an etiology is more often found in these idiopathic cases.
Cystic fibrosis (CF) is commonly associated with diffuse bronchiectasis, and previously undiagnosed CF may account for up to 20% of idiopathic cases. Even patients who are heterozygous, who typically have no clinical manifestations of CF, may have an increased risk of bronchiectasis.
Immunodeficiencies such as common variable immunodeficiency (CVID) may also lead to diffuse disease. Human immunodeficiency virus (HIV) infection and undernutrition also appear to increase risk.
Rare abnormalities in airway structure may lead to diffuse bronchiectasis.
Congenital defects in mucociliary clearance such as primary ciliary dyskinesia (PCD) syndromes may also be a cause, explaining almost 3% of previously idiopathic cases.
Diffuse bronchiectasis sometimes complicates common autoimmune disorders, such as rheumatoid arthritis or Sjögren syndrome, and can occur in the setting of hematologic malignancy, organ transplant, or due to the immunocompromise associated with treatment in these conditions. Bronchiectasis can also be related to more common conditions, including chronic obstructive pulmonary disease (COPD), asthma, or chronic, recurrent aspiration.
Allergic bronchopulmonary aspergillosis, a hypersensitivity reaction to Aspergillus species that occurs most commonly in people with asthma, but sometimes in patients with CF, can cause or contribute to bronchiectasis.
In countries where tuberculosis is common, most cases are probably caused by tuberculosis, particularly in patients with impaired immune function due to undernutrition or human immunodeficiency virus (HIV) infection.
Focal bronchiectasis
Focal bronchiectasis typically develops as a result of untreated pneumonia or obstruction (eg, due to foreign bodies, tumors, postsurgical changes, lymphadenopathy). Mycobacteria (tuberculous or nontuberculous) can both cause focal bronchiectasis and colonize the lungs of patients with bronchiectasis due to other disorders (see table Factors Predisposing to Bronchiectasis).
Pathophysiology of Bronchiectasis
The pathophysiology of bronchiectasis is not fully understood, likely in part because it is the common end-point of a heterogenous group of disorders predisposing to chronic airway inflammation. The most widely accepted model describes a "vicious cycle" of inflammation, airway destruction, abnormal mucus clearance, and infection or colonization by bacteria (1).
Diffuse bronchiectasis occurs when a causative disorder triggers inflammation of small and medium-sized airways, releasing inflammatory mediators from intraluminal neutrophils. The inflammatory mediators destroy elastin, cartilage, and muscle in larger airways, resulting in irreversible bronchodilation. Simultaneously, in the inflamed small and medium-sized airways, macrophages and lymphocytes form infiltrates that thicken mucosal walls. This thickening causes the airway obstruction frequently noted during pulmonary function testing.
With disease progression, inflammation spreads beyond the airways, causing fibrosis of the surrounding lung parenchyma. What inflames the small airways depends on the etiology of bronchiectasis. Common contributors include impaired airway clearance (due to production of thick, viscous mucus in CF, lack of ciliary motility in primary ciliary dyskinesia [PCD], or damage to the cilia and/or airways secondary to infection or injury) and impaired host defenses; these factors predispose patients to chronic infection and inflammation. In the case of immune deficiency (particularly CVID) and autoimmune causes, autoimmune inflammation may also contribute.
Focal bronchiectasis usually occurs when a large airway becomes obstructed. The resulting inability to clear secretions leads to a cycle of infection, inflammation, and airway wall damage. The right middle lobe is involved most often because its bronchus is small and angulated and has lymph nodes in close proximity. Lymphadenopathy due to mycobacterial infection sometimes causes bronchial obstruction and focal bronchiectasis.
As ongoing inflammation changes airway anatomy, pathogenic bacteria (sometimes including mycobacteria), colonize the airways. Common organisms include
Haemophilus influenzae
Moraxella catarrhalis
Pseudomonas aeruginosa
Staphylococcus aureus
Streptococcus pneumoniae
Nontuberculous mycobacteria
S. aureus colonization is strongly associated with CF; a culture finding of S. aureus should raise concern for undiagnosed CF. Also, colonization with P. aeruginosa tends to indicate severe disease and predicts worse outcomes, including increased risk of exacerbations, hospitalization, poor quality of life, rapid decline in lung function, and death. Colonization by multiple organisms is common, and antibiotic resistance is a concern in patients who require frequent courses of antibiotics for treatment of exacerbations.
Complications
As the disease progresses, chronic inflammation and hypoxemia cause neovascularization of the bronchial (not the pulmonary) arteries. Bronchial artery walls rupture easily, leading to hemoptysis, which can be massive and life threatening. Other vascular complications include pulmonary hypertension due to vasoconstriction, arteritis, and sometimes shunt from bronchial to pulmonary vessels.
Colonization with multidrug-resistant organisms can lead to chronic, low grade airway inflammation. This inflammation can progress, causing recurrent exacerbations and worsen airflow limitation on pulmonary function tests.
Pathophysiology reference
1. Cole PJ: Inflammation: a two-edged sword—the model of bronchiectasis. Eur J Respir Dis Suppl 147:6–15, 1986.
Symptoms and Signs of Bronchiectasis
Symptoms characteristically begin insidiously and gradually worsen over years, accompanied by episodes of acute exacerbation.
The most common presenting symptom is chronic cough that produces thick, tenacious, often purulent sputum. Dyspnea and wheezing are common, and pleuritic chest pain can develop. In advanced cases, hypoxemia and right-sided heart failure due to pulmonary hypertension may increase dyspnea. Hemoptysis, which can be massive, occurs due to airway neovascularization.
Acute exacerbations are common and frequently result from new or worsened infection. Exacerbations are marked by a worsening cough and increases in dyspnea and the volume and purulence of sputum. Low-grade fever and constitutional symptoms (eg, fatigue, malaise) may also be present.
Halitosis and abnormal breath sounds, including crackles, rhonchi, and wheezing, are typical physical examination findings. Digital clubbing is uncommon but may be present. Chronic rhinosinusitis and nasal polyps may be present, particularly in patients with CF or primary ciliary dyskinesia. Lean body mass commonly decreases, possibly due to inflammation and cytokine excess and, in patients with CF, malabsorption.
Diagnosis of Bronchiectasis
Chest x-ray
High-resolution chest CT
Pulmonary function tests for baseline evaluation and monitoring disease progression
Sputum culture for bacteria and mycobacteria to determine colonizing organisms
Specific tests for suspected causes
Diagnosis is based on history, physical examination, and radiologic testing, beginning with a chest x-ray. Chronic bronchitis may mimic bronchiectasis clinically, but bronchiectasis is distinguished by increased purulence and volume of daily sputum and by dilated airways shown on imaging studies.
Imaging
Chest x-ray is usually abnormal and may be diagnostic. X-ray findings suggestive of bronchiectasis involve thickening of the airway walls and/or airway dilation; typical findings include ill-defined linear perihilar densities with indistinctness of the central pulmonary arteries, indistinct rings due to thickened airways seen in cross section (parallel to the x-ray beam), and “tram lines” (or tram-track sign) caused by thickened, dilated airways perpendicular to the x-ray beam. Dilated airways filled with mucous plugs can also cause scattered elongated, tubular opacities.
Radiographic patterns may differ depending on the underlying disease; bronchiectasis due to cystic fibrosis develops predominantly in the upper lobes, whereas bronchiectasis due to an endobronchial obstruction causes more focal x-ray abnormalities.
High-resolution computed tomography (CT) is the test of choice for defining the extent of bronchiectasis, and is very sensitive and specific. Typical CT findings include airway dilation (in which the inner lumen of 2 or more airways exceed the diameter of the adjacent artery) and the signet ring sign, in which a thickened, dilated airway appears adjacent to a smaller artery in transaxial view. Lack of normal bronchial tapering can result in visible medium-sized bronchi extending almost to the pleura. "Tram lines" are easily visible on CT.
As airway damage increases over time, bronchiectasis changes progress from cylindrical to varicose and then cystic findings on imaging. Atelectasis, consolidation, mucous plugs, and decreased vascularity are nonspecific findings. In traction bronchiectasis, pulmonary fibrosis pulls or distorts airways in ways that simulate bronchiectasis on imaging.

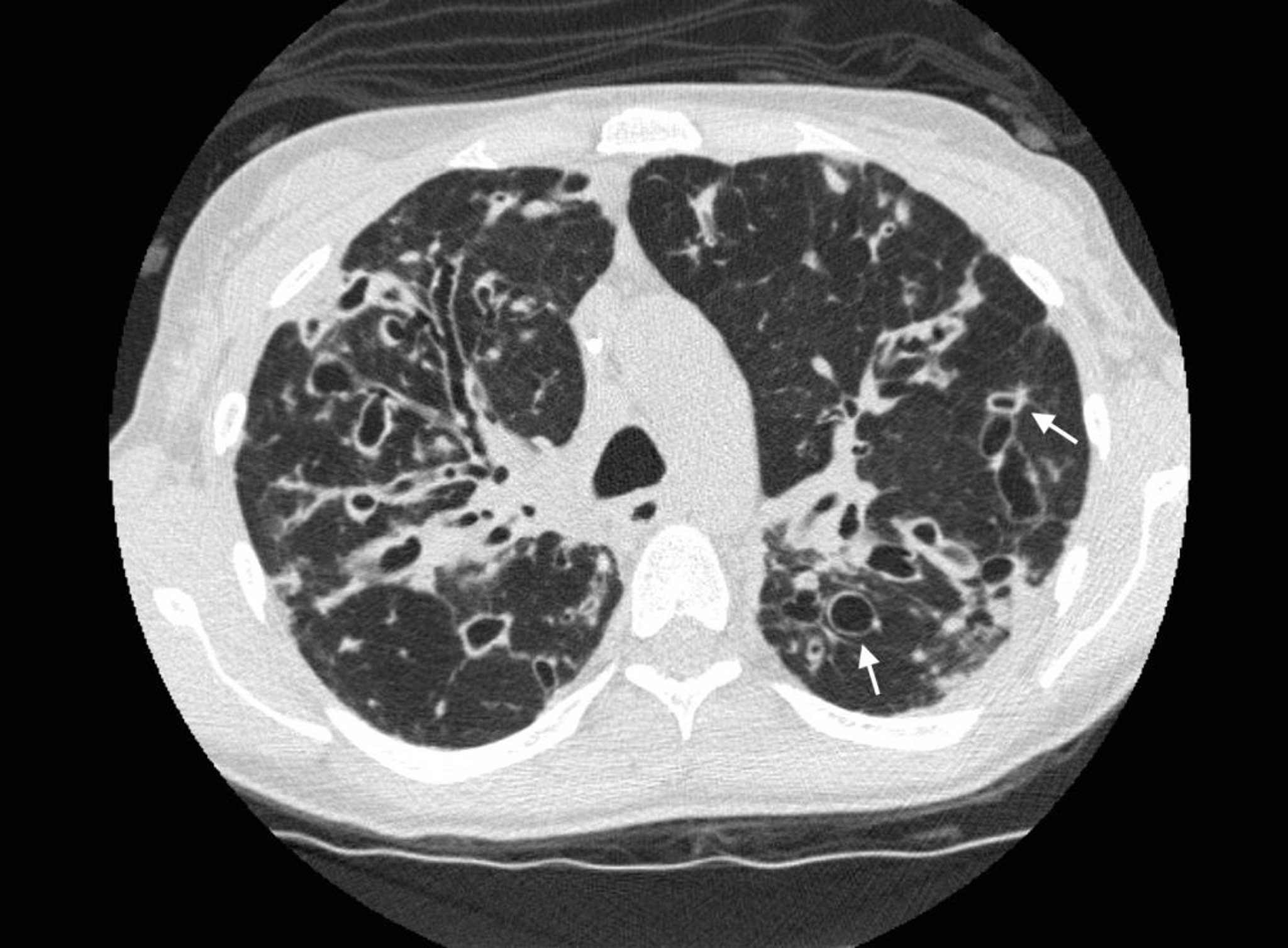
Photo courtesy of Başak Çoruh, MD.

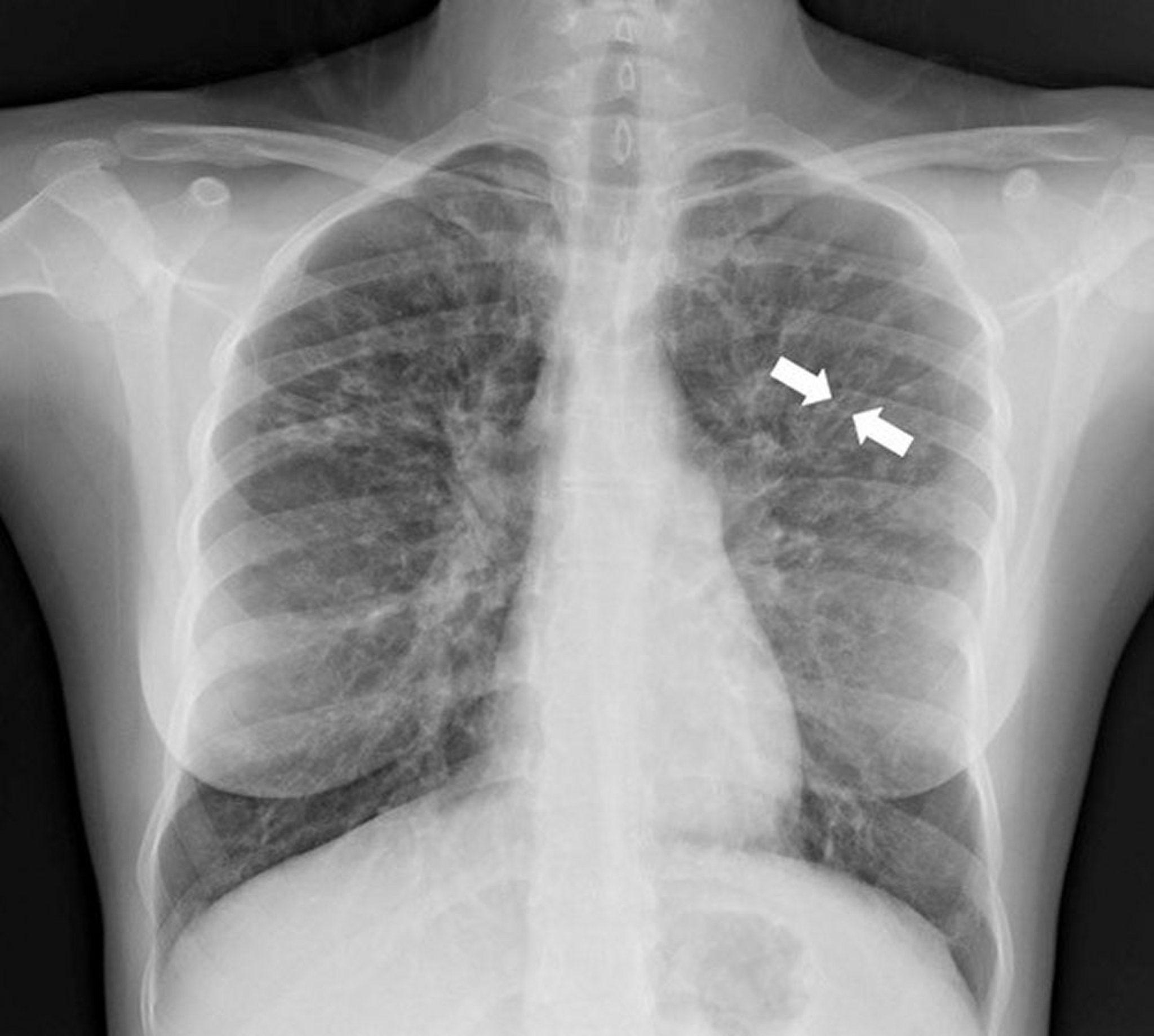
Photo courtesy of Başak Çoruh, MD.
Pulmonary function tests
Pulmonary function tests can be helpful for documenting baseline function and for monitoring disease progression. Bronchiectasis causes airflow limitation (reduced forced expiratory volume in 1 second [FEV1] with reduction in the FEV1/FVC ratio); the FEV1 may improve in response to beta-agonist bronchodilators. In more advanced cases, progressive fibrosis may result in decreases in forced vital capacity (FVC), evidence of a restrictive defect on lung volume measurements, and a decreased diffusing capacity for carbon monoxide (DLCO).
Diagnosis of cause
During an exacerbation-free period, all patients should have expectorated or induced sputum cultured to determine the predominant colonizing bacteria and their sensitivities. This information helps with antibiotic selection during exacerbations.
A complete blood count (CBC) and differential can help determine the severity of disease activity and identify eosinophilia, which may suggest complicating diagnoses.
Staining and cultures of sputum for bacterial, mycobacterial (Mycobacterium avium complex and M. tuberculosis), and fungal (Aspergillus species) organisms may also help identify the cause of chronic airway inflammation.
Clinically significant nontuberculous mycobacterial infection is diagnosed by finding high colony counts of these mycobacteria in cultures from serial sputum samples or from bronchoalveolar lavage fluid in patients who have granulomas on biopsy or concurrent radiologic evidence of disease.
When the cause of bronchiectasis is unclear, additional testing based on the history and imaging findings may be done. Tests may include the following:
Alpha-1 antitrypsin level to evaluate for alpha-1 antitrypsin deficiency if high-resolution CT shows lower lobe emphysema
Rheumatoid factor, antinuclear antibody (ANA), and antineutrophil cytoplasmic antibody testing if an autoimmune disorder is being considered
Serum immunoglobulins (IgG, IgA, IgM) and serum electrophoresis to diagnose common variable immunodeficiency
Serum IgE levels and Aspergillus precipitins if patients have eosinophilia, to rule out allergic bronchopulmonary aspergillosis
Sweat chloride tests (a positive test should be confirmed with a repeat test) and CFTR gene mutation analysis to diagnose cystic fibrosis (including in adults > 40 years without an identifiable cause of bronchiectasis, especially those with upper lobe involvement, malabsorption, or male infertility)
Patients who have laboratory evidence of immunodeficiency should be referred for to an immunology specialist for evaluation because test results are often challenging to interpret. Additional specialized testing may also be required to confirm the type of immunodeficiency present and determine treatment options.
Primary ciliary dyskinesia should be considered if adults with bronchiectasis also have chronic sinus disease or otitis media, particularly if problems have persisted since childhood. Bronchiectasis in such patients may have right middle lobe and lingular predominance, and infertility in males or dextrocardia may be present. Nasal or oral exhaled nitric oxide level is frequently low. Definitive diagnosis requires examination of a nasal or bronchial epithelial sample for abnormal ciliary structure using transmission electron microscopy.
The diagnosis of PCD should typically be done in specialized centers because evaluation can be challenging. Nonspecific structural defects can be present in up to 10% of cilia in healthy people and in patients with pulmonary disease, and infection can cause transient dyskinesia. Ciliary ultrastructure may also be normal in some patients with PCD syndromes, requiring further testing to identify abnormal ciliary function.
Bronchoscopy is indicated when an anatomic or obstructive lesion is suspected.
Definition and evaluation of exacerbations
A bronchiectasis exacerbation is defined as a patient with bronchiectasis with deterioration for at least 48 hours in ≥ 3 of the following symptoms (1):
Breathlessness and/or exercise intolerance
Cough
Fatigue and/or malaise
Hemoptysis
Sputum purulence
Sputum volume and/or consistency
The degree of testing depends on the severity of the clinical presentation. For patients with mild to moderate exacerbations, repeat sputum cultures to confirm the causative organism and sensitivity patterns may be sufficient. These tests help narrow antibiotic coverage and exclude opportunistic pathogens.
For more severely ill patients, a CBC, chest x-ray, and possibly other tests may be warranted to exclude common complications of serious pulmonary infection, such as lung abscess and empyema.
Diagnosis reference
1. Hill AT, Haworth CS, Aliberti S, et al: Pulmonary exacerbation in adults with bronchiectasis: A consensus definition for clinical research. Eur Respir J 49:1700051, 2017.
Treatment of Bronchiectasis
Prevention of exacerbations with regular vaccinations and sometimes suppressive antibiotics
Measures to help clear airway secretions
Bronchodilators and sometimes inhaled corticosteroids if reversible airway obstruction is present
Antibiotics and bronchodilators for acute exacerbations
Early treatment with antivirals of any viral infections, particularly influenza and COVID-19
Sometimes surgical resection for localized disease with intractable symptoms or bleeding
Lung transplantation in carefully selected patients who have advanced disease despite maximal therapy
The key treatment goals are to control symptoms and improve quality of life, reduce the frequency of exacerbations, and preserve lung function (1, 2).
As for all patients with chronic pulmonary disease, recommendations include the following:
Airway clearance techniques are used to reduce chronic cough in patients with significant sputum production and mucous plugging and to reduce symptoms during exacerbations. Such techniques include regular exercise, chest physiotherapy with postural drainage and chest percussion, positive expiratory pressure devices, intrapulmonary percussive ventilators, pneumatic vests, and autogenic drainage (a breathing technique thought to help move secretions from peripheral to central airways). Patients should be taught these techniques by a respiratory therapist and should use whichever one is most effective and sustainable for them; no evidence favors one particular technique. Patients should be advised to continue their airway clearance techniques for at least 10 minutes and may stop when they produce 2 clear coughs or huffs (3) or 30 minutes has passed.
For patients with reversible airway obstruction, bronchodilator therapy (eg, with some combination of a long-acting beta-adrenergic agonist, a long-acting muscarinic antagonist, and a short-acting beta-adrenergic drug as indicated by symptoms and severity of lung obstruction, as used in patients with COPD) can help improve lung function and quality of life. Inhaled corticosteroids may also be used in patients with frequent exacerbations or marked variability in lung function measurements (ie, reversible airway obstruction following bronchodilator administration), but their role remains controversial. Pulmonary rehabilitation can be helpful. Maintaining adequate hydration is also important.
There is no consensus on the best use of antibiotics to prevent or limit the frequency of acute exacerbations. Use of suppressive antibiotics regularly or on a rotating schedule reduces symptoms and exacerbations but may increase the risk that future infections will involve resistant organisms. Current guidelines suggest using antibiotics in patients with ≥ 3 exacerbations per year and possibly also in those with fewer exacerbations who have culture-proven P. aeruginosa colonization. Some guidelines suggest attempting eradication of organisms such as P. aeruginosa or S. aureus when they are first detected in sputum cultures (3).
Chronic macrolide therapy reduces acute exacerbations in patients with bronchiectasis, and can slow the decline in lung function in patients with CF (4, 5, 6). Macrolides are thought to be beneficial mainly due to their anti-inflammatory or immunomodulatory effects.
Treatment sessions for bronchiectasis should be done in the following order:
Inhaled short-acting bronchodilators
Mucolytic therapy (if prescribed)
Airway clearance technique
Any prescribed inhaled or nebulized antibiotics, long-acting bronchodilators, or corticosteroids
Underlying conditions should be treated to slow the progression of lung disease.
For patients with underlying immunodeficiency states: Scheduled intravenous immunoglobulin (which may reduce the frequency of lower respiratory infections [7])
For patients with cystic fibrosis: Antibiotics and inhaled bronchodilators as well as comprehensive support, and dietary supplementation. Most patients with cystic fibrosis benefit from CFTR modulator therapy, which can decrease exacerbations. Patients with CF should receive all or part of their care by teams with expertise in CF, generally at a designated CF care center.
For patients with allergic bronchopulmonary aspergillosis: Corticosteroids and sometimes azole antifungals
For patients with alpha-1 antitrypsin deficiency: Replacement therapy
Acute exacerbations
Acute exacerbations are treated with antibiotics, inhaled bronchodilators (particularly if patients are wheezing), and increased attempts at mucus clearance, using mechanical techniques, treatment of dehydration (if present), humidification, and nebulized saline (and mucolytics for patients with CF). Inhaled or oral corticosteroids are frequently given to treat airway inflammation and worsening airway obstruction. Antibiotic choice depends on previous culture results and whether or not patients have CF (8).
Initial antibiotics for patients without CF and with no prior culture results should be effective against H. influenzae, M. catarrhalis, S. aureus, and S. pneumoniaeP. aeruginosaP. aeruginosa is detected. Shorter courses are reserved for patients with mild disease.
Initial antibiotic selection for patients with CF is guided by previous sputum culture results (done routinely in all patients with CF). During childhood, common infecting organisms are S. aureus and H. influenzaeP. aeruginosa, Burkholderia cepacia, and Stenotrophomonas maltophilia
Complications
Significant hemoptysis is usually treated with bronchial artery embolization, but surgical resection may be considered if embolization is ineffective and pulmonary function is adequate.
Superinfection with mycobacterial organisms such as M. avium
Surgical resection is rarely indicated but may be considered when bronchiectasis is localized, medical therapy has been optimized, and the symptoms are intolerable. In certain patients with diffuse bronchiectasis, especially cystic fibrosis, lung transplantation is also an option.
Treatment references
1. Polverino E, Gemine PC, McDonnell MJ, et al: European Respiratory Society guidelines for the management of adult bronchiectasis. Eur Respir J 50: 1700629, 2017. doi: 10.1183/13993003.00629-2017
2. Nicholson CH, Holland AE, Lee AL: The Bronchiectasis Toolbox - A Comprehensive Website for the Management of People with Bronchiectasis. Med Sci (Basel) 5, 13, 2017.
3. Hill AT, Sullivan AL, Chalmers JD, et al: British Thoracic Society Guideline for bronchiectasis in adults. Thorax 74(Suppl 1):1–69, 2019. doi: 10.1136/thoraxjnl-2018-212463
4. Wong C, Jayaram L, Kraals N, et al: Azithromycin for the prevention of exacerbations in non-cystic fibrosis bronchiectasis (EMBRACE): A randomised, double blind, placebo controlled trial. Lancet 380: 660–667, 2012.
5. Altenburg J, de Graaf CS, Stienetra Y, et al: Effect of azithromycin maintenance treatment on infectious exacerbations among patients with non-cystic fibrosis bronchiectasis: The BAT randomized controlled trial. JAMA 309: 1251–1259, 2013.
6. Serisier DJ, Martin ML, McGuckin MA, et al: Effect of long-term, low dose erythromycin on pulmonary exacerbations among patients with non-cystic fibrosis bronchiectasis: the BLESS randomized controlled trial. JAMA 309: 1260–1267, 2013.
7. Quinti I, Sorellina A, Guerra A, et al: Effectiveness of immunoglobulin replacement therapy on clinical outcome in patients with primary antibody deficiencies: Results from a multicenter prospective cohort trial. J Clin Immunol 31: 315–322, 2011.
8. Flume PA, Mogayzel PJ Jr, Robinson KA, et al: Cystic fibrosis pulmonary guidelines: Treatment of pulmonary exacerbations. Am J Respir Crit Care Med 80:802–808, 2009. doi: 10.1164/rccm.200812-1845PP
Prognosis for Bronchiectasis
Prognosis varies widely. Mean yearly decrease in FEV1 is about 50 to 55 mL (normal decrease in healthy people is about 20 to 30 mL). Patients with CF historically have had the poorest prognosis, with a median survival of 36 years. However, the advent of CFTR (cystic fibrosis transmembrane regulator) modulator therapy has resulted in meaningful improvements in outcomes, even in patients with advanced lung disease (1).
Prognosis reference
1. Shteinberg M, Taylor-Cousar JL: Impact of CFTR modulator use on outcomes in people with severe cystic fibrosis lung disease. Eur Respir Rev 29(155):190112, 2020. doi: 10.1183/16000617.0112-2019
Key Points
In bronchiectasis, chronic inflammation from various causes destroys elastin, cartilage, and muscle in larger airways, resulting in irreversible damage and dilated airways that are chronically colonized by infectious organisms.
Patients have chronic productive cough with intermittent acute exacerbations.
Diagnosis is with imaging, usually CT; cultures should be done to identify colonizing organism(s).
Prevent exacerbations using appropriate immunizations, airway clearance measures, and sometimes macrolide antibiotics.
Treat exacerbations with antibiotics, bronchodilators, more frequent airway clearance measures, and sometimes corticosteroids.






