


Ventricular tachycardia is 3 consecutive ventricular beats at a rate 120 beats/minute. Symptoms depend on duration and vary from none to palpitations to hemodynamic collapse and death. Diagnosis is by electrocardiography. Treatment of more than brief episodes is with cardioversion or antiarrhythmics, depending on symptoms. If necessary, long-term treatment is with an implantable cardioverter defibrillator.
(See also Overview of Arrhythmias.)
Some experts use a cutoff rate of ≥ 100 beats/minute for ventricular tachycardia (VT). Repetitive ventricular rhythms at slower rates are called accelerated idioventricular rhythms or slow VT; they are usually benign and are not treated unless patients have hemodynamic symptoms.
Most patients with VT have a significant heart disorder, particularly prior myocardial infarction or a cardiomyopathy. Electrolyte abnormalities (particularly hypokalemia or hypomagnesemia), acidemia, hypoxemia, and adverse drug effects contribute. The long QT syndrome (congenital or acquired) is associated with a particular form of VT, torsades de pointes.
Ventricular tachycardia may be monomorphic or polymorphic and nonsustained or sustained.
Monomorphic VT: Single abnormal focus or reentrant pathway and thus regular, identical-appearing QRS complexes
Polymorphic VT: Several different foci or pathways and thus irregular, varying QRS complexes
Nonsustained VT: Lasts < 30 seconds
Sustained VT: Lasts ≥ 30 seconds or is terminated sooner because of hemodynamic collapse
Catecholaminergic polymorphic ventricular tachycardia is a genetic disorder affecting intracellular calcium regulation in cardiac tissue. Patients are predisposed to atrial and/or ventricular tachyarrhythmias and sudden cardiac death, particularly during increased adrenergic activity.
VT frequently deteriorates to ventricular fibrillation and thus cardiac arrest.
Symptoms and Signs of Ventricular Tachycardia
Ventricular tachycardia of short duration or slow rate may be asymptomatic. Sustained VT is almost always symptomatic, causing palpitations, symptoms of hemodynamic compromise, or sudden cardiac death.
Diagnosis of Ventricular Tachycardia
Electrocardiography (ECG)
Diagnosis of ventricular tachycardia is by ECG (see figure Broad QRS ventricular tachycardia). Any wide QRS complex tachycardia (QRS ≥ 0.12 second) should be considered VT until proved otherwise.
Pearls & Pitfalls
|
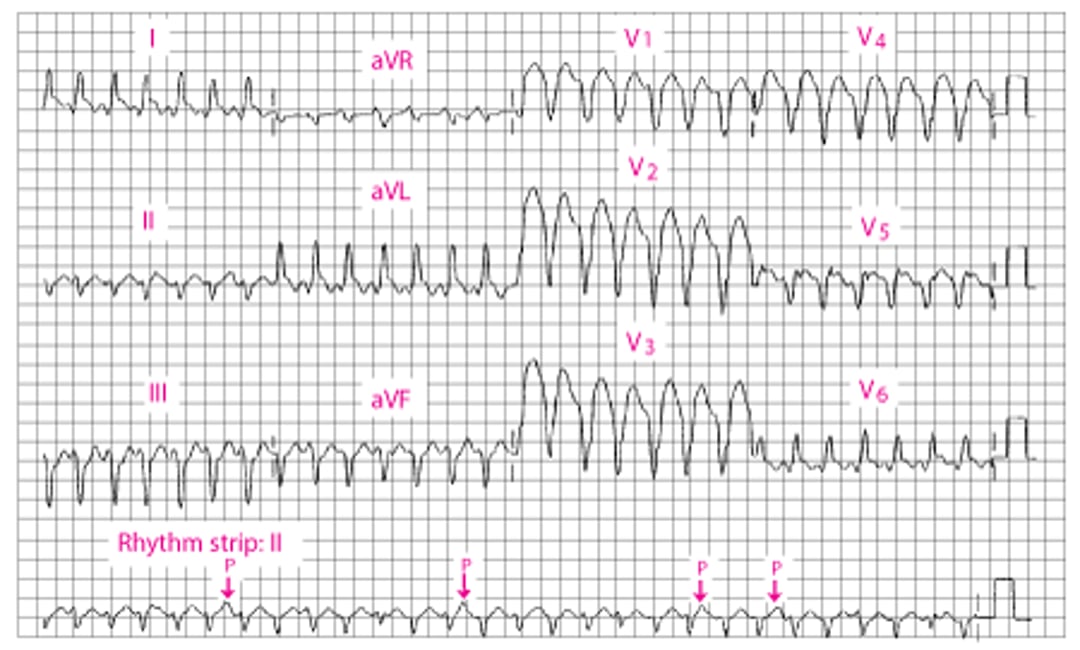
Diagnosis is supported by ECG findings of dissociated P-wave activity, fusion or capture beats, uniformity of QRS vectors in the V leads (concordance) with discordant T-wave vector (opposite QRS vectors), and a frontal-plane QRS axis in the northwest quadrant. Differential diagnosis includes supraventricular tachycardia conducted with bundle branch block or via an accessory pathway (see figure Modified Brugada Criteria for ventricular tachycardia
Pearls & Pitfalls
|
Broad QRS Ventricular Tachycardia
The QRS duration is 160 millisecond. An independent P wave can be seen in II (arrows). There is a leftward mean frontal axis shift. |

Treatment of Ventricular Tachycardia
Acute: Sometimes synchronized direct current cardioversion, sometimes class I or class III antiarrhythmics
Long-term: Usually an implantable cardioverter-defibrillator
Acute
Treatment of acute ventricular tachycardia depends on symptoms and duration of VT.
Pulseless VT requires defibrillation with ≥100 joules.
Stable sustained VT can be treated with synchronized direct current cardioversion with ≥100 joules.
Stable sustained VT can also be treated with intravenous class I or class III antiarrhythmic drugs (see table Antiarrhythmic Drugs
Nonsustained VT does not require immediate treatment unless the runs are frequent or long enough to cause symptoms. In such cases, antiarrhythmics are used as for sustained VT.
Long-term
The primary goal is preventing sudden death, rather than simply suppressing the arrhythmia. It is best accomplished by use of an implantable cardioverter-defibrillator (ICD). However, the decision about whom to treat is complex and depends on the estimated probability of life-threatening VTs and the severity of underlying heart disorders (see table Indications for Implantable Cardioverter-Defibrillators).
Long-term treatment is not required when the index episode of ventricular tachycardia resulted from a transient cause (eg, during the 48 hours after onset of myocardial infarction) or a reversible cause (acid-base disturbances, electrolyte abnormalities, proarrhythmic drug effect).
Because nonsustained VT is a marker for increased risk of sudden death in patients with a structural heart disorder, such patients (particularly those with an ejection fraction < 0.35) require further evaluation. Such patients should receive an ICD.
When prevention of VTs is important (usually in patients who have an ICD and are having frequent episodes of VT), antiarrhythmics or transcatheter or surgical ablation of the arrhythmogenic substrate is required. Any class Ia, Ib, Ic, II, or III antiarrhythmic drug
Transcatheter ablation
Key Points
Any wide-complex (QRS ≥ 0.12 second) tachycardia should be considered ventricular tachycardia (VT) until proved otherwise.
Patients who do not have a pulse should be cardioverted.
Synchronized cardioversion or antiarrhythmic drugs may be tried if the patient is stable.
Patients who had an episode of sustained VT without a transient or reversible cause typically require an implantable cardioverter-defibrillator (ICD).






