


Tubulointerstitial nephritis is primary injury to renal tubules and interstitium resulting in decreased renal function. The acute form is most often due to allergic drug reactions or to infections. The chronic form occurs with a diverse array of causes, including genetic or metabolic disorders, obstructive uropathy, and chronic exposure to environmental toxins or to certain medications and herbs. Diagnosis is suggested by history and urinalysis and often confirmed by biopsy. Treatment and prognosis vary by the etiology and potential reversibility of the disorder at the time of diagnosis.
(See also Overview of Tubulointerstitial Diseases.)
Etiology of Tubulointerstitial Nephritis
Tubulointerstitial nephritis can be primary, but a similar process can result from glomerular damage or renovascular disorders.
Primary tubulointerstitial nephritis may be
Acute (see table Causes of Acute Tubulointerstitial Nephritis)
Chronic (see table Causes of Chronic Tubulointerstitial Nephritis)
Acute tubulointerstitial nephritis
Acute tubulointerstitial nephritis (ATIN) involves an inflammatory infiltrate and edema affecting the renal interstitium that often develops over days to months. Over 95% of cases result from infection or an allergic drug reaction.
ATIN causes acute kidney injury; severe cases, delayed therapy, or continuance of an offending medication can lead to permanent injury and chronic kidney disease.
Renal-ocular syndrome, acute tubulointerstitial nephritis plus uveitis, also occurs and is idiopathic.
Chronic tubulointerstitial nephritis
Chronic tubulointerstitial nephritis (CTIN) arises when chronic tubular insults cause gradual interstitial infiltration and fibrosis, tubular atrophy and dysfunction, and a gradual deterioration of renal function, usually over years. Concurrent glomerular involvement (glomerulosclerosis) is much more common in CTIN than in ATIN.
Causes of chronic tubulointerstitial nephritis are myriad; they include immunologically mediated disorders, infections, reflux or obstructive nephropathy, medications, and other disorders. CTIN due to toxins, metabolic derangements, hypertension, and inherited disorders results in symmetric and bilateral disease; when CTIN is due to other causes, renal scarring may be unequal and involve only one kidney. Some well-characterized forms of CTIN include
Reflux nephropathy and myeloma may cause tubulointerstitial injury but the predominant pathology in these conditions is glomerular disease.
Hereditary cystic kidney diseases are discussed elsewhere.
General references
1. Yamaguchi Y, Kanetsuna Y, Honda K, et al: Characteristic tubulointerstitial nephritis in IgG4-related disease. Hum Pathol 43(4):536-549, 2012. doi: 10.1016/j.humpath.2011.06.002
2. Correa-Rotter R, Wesseling C, Johnson RJ: CKD of unknown origin in Central America: The case for Mesoamerican nephropathy. Am J Kidney Dis 63(3):506-520, 2014. doi: 10.1053/j.ajkd.2013.10.062
Symptoms and Signs of Tubulointerstitial Nephritis
Acute tubulointerstitial nephritis
Symptoms and signs of acute tubulointerstitial nephritis (ATIN) may be nonspecific and are often absent unless symptoms and signs of renal failure develop. Many patients develop polyuria and nocturia (due to a defect in urinary concentration and sodium reabsorption).
< 10% of patients with medication-induced ATIN. Abdominal pain, weight loss, and bilateral renal enlargement (caused by interstitial edema) may also occur in ATIN and with fever may mistakenly suggest renal cancer or polycystic kidney disease. Peripheral edema and hypertension are uncommon unless renal failure occurs.
Chronic tubulointerstitial nephritis
Symptoms and signs are generally absent in chronic tubulointerstitial nephritis unless renal failure develops. Edema usually is not present, and blood pressure is normal or only mildly elevated in the early stages. Polyuria and nocturia may develop.
Diagnosis of Tubulointerstitial Nephritis
Risk factors
In acute tubulointerstitial nephritis (ATIN), active urinary sediment with sterile pyuria
Sometimes renal biopsy
Usually imaging to exclude other causes
Few clinical and routine laboratory findings are specific for tubulointerstitial nephritis. Thus, suspicion should be high when the following are present:
Typical symptoms or signs
Risk factors, particularly a temporal relationship between onset and use of a potentially causative medication
Characteristic urinalysis findings, particularly sterile pyuria
Modest proteinuria, usually < 1 g/day (except with use of nonsteroidal anti-inflammatory drugs [NSAIDs], which may cause nephrotic-range proteinuria, 3.5 g/day)
Evidence of tubular dysfunction (eg, renal tubular acidosis, Fanconi syndrome)
Concentrating defect out of proportion to the degree of renal failure
Eosinophiluria cannot be relied upon to make or exclude the diagnosis. Other tests (eg, imaging) are usually necessary to differentiate ATIN or chronic tubulointerstitial nephritis (CTIN) from other disorders. A presumptive clinical diagnosis of ATIN is often made based on the specific findings mentioned above, but renal biopsy is necessary to establish a definitive diagnosis.
Acute tubulointerstitial nephritis
Urinalysis
Blood test findings of tubular dysfunction include hypokalemia (caused by a defect in potassium reabsorption) and a nonanion gap metabolic acidosis (caused by a defect in proximal tubular bicarbonate reabsorption or in distal tubular acid excretion). Elevated serum total immunoglobulin G (IgG) and/or IgG4 levels and low serum complement concentrations (ie, low C3 and C4) may be present in patients with IgG4-related disease or hypocomplementemic interstitial nephritis.
Ultrasonography, radionuclide scanning, or both may be needed to differentiate acute tubulointerstitial nephritis from other causes of acute kidney injury when kidney biopsy is not possible. In ATIN, ultrasonography may show kidneys that are greatly enlarged and echogenic because of interstitial inflammatory cells and edema. Radionuclide scans may show kidneys avidly taking up radioactive gallium-67 or radionuclide-labeled white blood cells (WBCs). Positive scans strongly suggest ATIN (and indicate that acute tubular necrosis is less likely), but a negative scan does not exclude ATIN.
Renal biopsy is usually reserved for patients with the following:
An uncertain diagnosis
Progressive renal injury
No improvement after potential causative medications are stopped
Findings suggesting early disease
Medication-induced ATIN for which corticosteroid therapy is under consideration
In acute tubulointerstitial nephritis, glomeruli are usually normal. The earliest finding is interstitial edema, typically followed by interstitial infiltration with lymphocytes, plasma cells, eosinophils, and a few polymorphonuclear leukocytes. In severe cases, inflammatory cells can be seen invading the space between the cells lining the tubular basement membrane (tubulitis); in other specimens, granulomatous reactions resulting from exposure to beta-lactam antibiotics, sulfonamides, mycobacteria, or fungi may be seen. The presence of noncaseating granulomas suggests sarcoidosis. A lymphoplasmacytic infiltration of the kidney interstitium with storiform fibrosis suggests IgG4-related tubulointerstitial nephritis. Immunofluorescence or electron microscopy seldom reveals any pathognomonic changes.

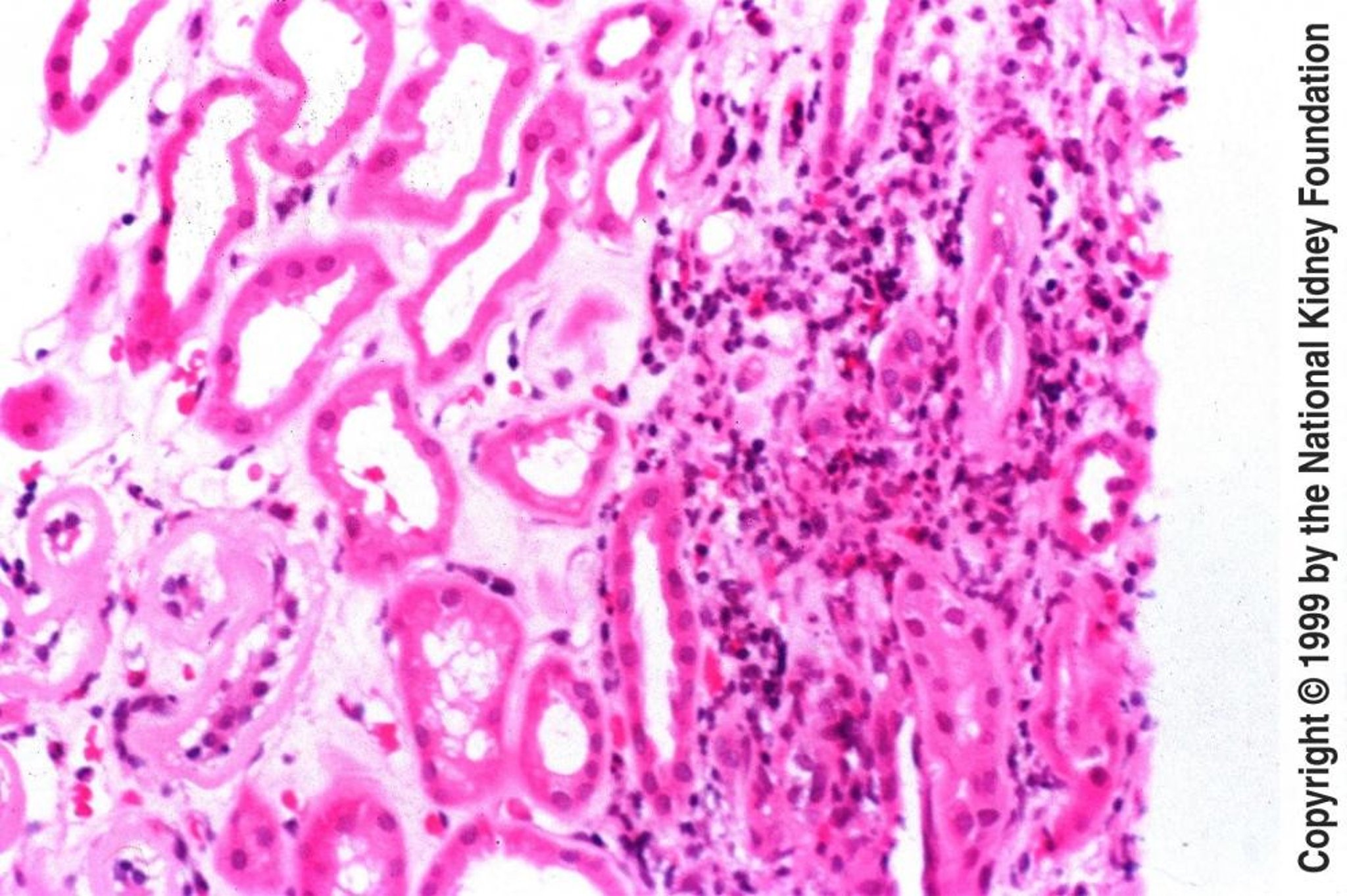
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
Chronic tubulointerstitial nephritis
Findings of CTIN are generally similar to those of ATIN, although urinary RBCs and WBCs are uncommon. Because CTIN is insidious in onset and interstitial fibrosis is common, imaging tests may show small kidneys with evidence of scarring and asymmetry.
In chronic tubulointerstitial nephritis, renal biopsy is not often done for diagnostic purposes. However, if there is concern for alternative diagnoses, it could be pursued. Glomeruli vary from normal to completely destroyed. Tubules may be absent or atrophied. Tubular lumina vary in diameter but may show marked dilation, with homogeneous casts. The interstitium contains varying degrees of inflammatory cells and fibrosis. Nonscarred areas appear almost normal. Grossly, the kidneys are small and atrophic.
Treatment of Tubulointerstitial Nephritis
Treatment of cause (eg, stopping the causative medication)
Corticosteroids for immune-mediated and sometimes medication-induced acute tubulointerstitial nephritis
Treatment of both ATIN and CTIN is management of the cause.
For medication-induced ATIN, corticosteroids are most effective when given within 2 weeks of stopping the causative medications. NSAID-induced ATIN is less responsive to corticosteroids than other medication-induced ATIN. ATIN should be proven by biopsy before corticosteroids are started.
Pearls & Pitfalls
Prognosis for Tubulointerstitial Nephritis
Acute tubulointerstitial nephritis
In medication-induced ATIN, renal function usually recovers within 6 to 8 weeks when the causative medication is stopped, although some residual scarring is common. Recovery may be incomplete, with persistent azotemia above baseline. Prognosis is usually worse if ATIN is caused by NSAIDs than by other medications. When other factors cause ATIN, histologic changes usually are reversible if the cause is recognized and removed; however, some severe cases progress to fibrosis and chronic kidney disease. Regardless of cause, irreversible injury is suggested by the following:
Diffuse rather than patchy interstitial infiltrate
Significant interstitial fibrosis
Acute kidney injury lasting >3 weeks
Chronic tubulointerstitial nephritis
In CTIN, prognosis depends on the cause and on the ability to recognize and stop the process before irreversible fibrosis occurs. Many genetic (eg, cystic kidney disease), metabolic (eg, cystinosis), and toxic (eg, previous exposure to heavy metals) causes may not be modifiable, in which case CTIN usually evolves to end-stage kidney disease.
Key Points
Causes of chronic tubulointerstitial nephritis (CTIN) are myriad and much more diverse than those for acute tubulointerstitial nephritis (ATIN), which is usually caused by an allergic reaction to a medication or by an infection.
Symptoms are often absent or nonspecific, particularly in CTIN.
Suspect the diagnosis based on risk factors and urinary sediment, exclude other causes using imaging, and sometimes confirm the diagnosis by biopsy.
Stop causative medications, treat any other causes, and provide supportive treatment.
Treat biopsy-proven immune-mediated and sometimes drug-induced ATIN with corticosteroids (within 2 weeks of stopping any causative medications).






